Rosenfeld Group
Liquid biopsies and cancer diagnostics

Research summary
Cancer patients’ blood contains DNA fragments from tumors. Using advanced genomic technologies, we have developed liquid biopsies to help diagnose cancer and allow us to study its evolution and treatment resistance noninvasively. This method could offer more detailed and frequent monitoring compared to traditional tissue biopsies, leading to better research and improved cancer care.

Professor Nitzan Rosenfeld
Senior Group Leader,
Director of Barts Cancer Institute
Research areas
Non-invasive diagnostic tools for cancer using circulating DNA
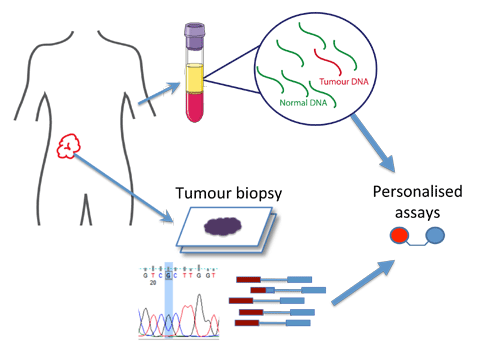
We employ emerging molecular technologies to develop new diagnostic approaches. Our focus is on circulating tumour DNA (ctDNA) as a noninvasive modality to assess evolution of solid malignancies. This is DNA originating from cancer cells, carrying tumour-specific genomic alterations, that is present as short cell-free fragments in body fluids such as blood plasma. ctDNA can be collected noninvasively via blood samples and has the potential to be immensely informative.
The field of prenatal diagnostics is revolutionised by non-invasive tests that assay fetal DNA fragments in maternal plasma. Parallel progress in cancer has been lagging, because genomic loci of interest are not well defined, and levels of tumour DNA in plasma are variable and generally lower: 2 ml of plasma may contain as many as 10,000 copies of DNA from healthy cells but only a few dozen copies of the tumour genome. The mechanisms through which tumour DNA reaches blood circulation are unclear, although fragmentation patterns of DNA in the plasma suggest it may originate from cell death. Overall levels of circulating DNA are higher in cancer patients compared with healthy controls, but these differences are not consistent enough for robust diagnostic tools. Maturation of genomic technologies empowers a different approach that focuses on those fragments that carry cancer mutations.
We use a combination of molecular methods such as next-generation sequencing and digital PCR, and develop bespoke data analysis algorithms that allow such data to be used for sensitive measurement of rare alleles. We apply these methods to monitor mutation status and circulating tumour DNA levels in serial samples collected from patients during treatment and follow-up, in close collaboration with clinical and translational research groups. High levels of circulating tumour DNA in cancer patients are a bad prognostic indicator. Changes in circulating tumour DNA levels may indicate response to therapy or disease progression, and indeed may prove to be the earliest indicator of changes to tumour burden (Dawson et al., N Engl J Med 2013; 368: 1199).
We have developed methods that allow us to use circulating tumour DNA as a “liquid biopsy” to analyse cancer mutations in a non-invasive way (Forshew et al., Sci Transl Med 2012; 4: 136ra68) (Figure 2). This can identify if a tumour is likely to respond to novel therapies that target specific cancer mutations or molecular pathways, through analysis of a blood test, and can reduce the dependence on obtaining a tumour biopsy which involves invasive procedures. This is especially helpful in clinical scenarios where obtaining a biopsy is not possible due to risks to the patient, but also opens up the possibility of repeated or serial analysis for such mutations during treatment and patient follow-up.
Non-invasive analysis of cancer evolution and resistance to therapy using circulating DNA
At present, a significant effort in cancer research is in understanding the roles of tumour heterogeneity and evolution on drug response and resistance. Much of this depends to date on collection of multiple biopsy samples from generous patients, which limits its applicability and adds complexity and costs. Together with the Caldas and Brenton Groups, we have recently shown that analysis of serial plasma samples can be used to identify mutations that are selected for when cancer relapses after therapy, and are therefore likely to contribute to resistance to the therapy (Murtaza et al., Nature 2013; 497: 108) (Figure 3). We are further developing these approaches, which can provide data in a non-invasive manner and can greatly enhance the pace of research.
Related News
See all news-

Prof Nitzan Rosenfeld appointed Director of Barts Cancer Institute
22nd September 2023
Professor Nitzan Rosenfeld has been appointed as Queen Mary University of London’s new Director of the Barts Cancer Institute.
Find out more -

Institute Group Leaders awarded promotions by the University
15th June 2022
We are delighted to announce that Florian Markowetz and Nitzan Rosenfeld have been promoted to Professors in the University of Cambridge Senior Academic Promotions.
Find out more -
Personalised blood test can detect persistent lung cancer
17th March 2022
Patients who are at a higher risk of their lung cancer returning can be identified by a personalised blood test that is performed after treatment, according to researchers at the University of Cambridge.
Find out more


Laboratory Efficiency Assessment Framework (LEAF)
The Rosenfeld Group contributed to the Institute’s LEAF Silver accreditation, see the Sustainability webpage for more information.