Carroll Group
Nuclear receptor transcription

Research summary
Our group studies how certain cancers grow and spread, especially those that depend on hormones. We focus on special proteins called transcription factors, which control genes by turning them on and off. These genes can tell cancer cells to keep growing. We want to find where these control points are and how these proteins interact with them so we can find ways to stop cancer growth.
Introduction
We are interested in defining the genomic and molecular features of transcription factors in cancer. We are specifically focused on identifying, characterising and modulating pioneer factors, a specialised class of transcription factors that create regulatory elements and initiate gene expression events. Pioneer factors are important in defining cell lineage and phenotype making them critical in cancer development and progression. The archetypal pioneer factor is a protein called FOXA1, which plays a critical role in the most common breast cancer subtype, Estrogen Receptor positive (ER+) breast cancer, as well as in prostate cancer. More recently, FOXA1 has been shown to play a role in other cancer types, including pancreatic cancer.
FOXA1 and other pioneer factors are able to open compacted chromatin, creating enhancer elements that can subsequently be used by nuclear receptor transcription factors, such as Estrogen Receptor in breast cancer and Androgen Receptor in prostate cancer. These nuclear receptors can then form the platform for recruitment of co-factors, many of which have enzymatic or structural roles required for the regulation of transcription.
We are interested in understanding how these nuclear receptors interact with the DNA, what the critical functional co-factors are and how changes in pioneer factor levels or fidelity can alter these processes. A major part of this is identifying where in the genome the transcription factors associate with the DNA and what the proteins are in the complexes at these binding sites. We use various genomic approaches and couple these with proteomic approaches to characterise these events. A method we developed called RIME (and the quantitative version called qPLEX-RIME) permits the discovery of protein complexes and is one of the only approaches for protein interactome discovery that is applicable for clinical tissue. The RIME approach enables the discovery of important protein complexes in tumour tissue, as well as being a fantastic tool for identifying how key protein complexes change from treatment sensitive to treatment resistant cancers, and from primary tumours to metastases.

Professor Jason Carroll
Interim Director and Senior Group Leader
Research topics
Identifying the proteins and perturbations that drive disease progression
We focus on characterising the events that culminate in ER-mediated gene expression. This involves mapping ER and co-factor binding sites from models and tumour tissue and defining the genomic properties that enable ER to mediate gene expression from distal enhancer elements. We have been particularly focused on mapping transcription factor binding sites from clinical tissue and we were one of the first labs to establish transcription factor mapping from tumour samples. We are specifically interested in identifying the key co-factors in ER+ breast cancer, with the goal of using small molecule inhibitors against these proteins, based on novel biological insight. We identify the genomic changes that can contribute to disease progression, including the identification of genes and proteins that mediate treatment resistance and metastasis. We use many approaches to define these events, including CRISPR screens, proteomics and genomics, which we integrate with data from clinical samples to inform us on candidates that are likely to be functionally important. We do deep functional characterisation of how identified molecular changes contribute to resistance and metastasis, in part by exploiting the physiologically-relevant intraductal system that, for the first time, accurately models ER+ breast cancer metastasis.
In addition to understanding how ER functions in ER+ breast cancer, we explore the substantial cross-talk that exists between ER and other nuclear receptors in breast cancer. Work from our group and our collaborators have shown that Progesterone Receptor and Androgen Receptor, both have the ability to impinge on ER activity by competing with ER for both DNA binding sites and rate-limiting co-factors. We use this information to inform clinical trials that exploit PR and AR ligands for therapeutic use in ER+ breast cancer patients.
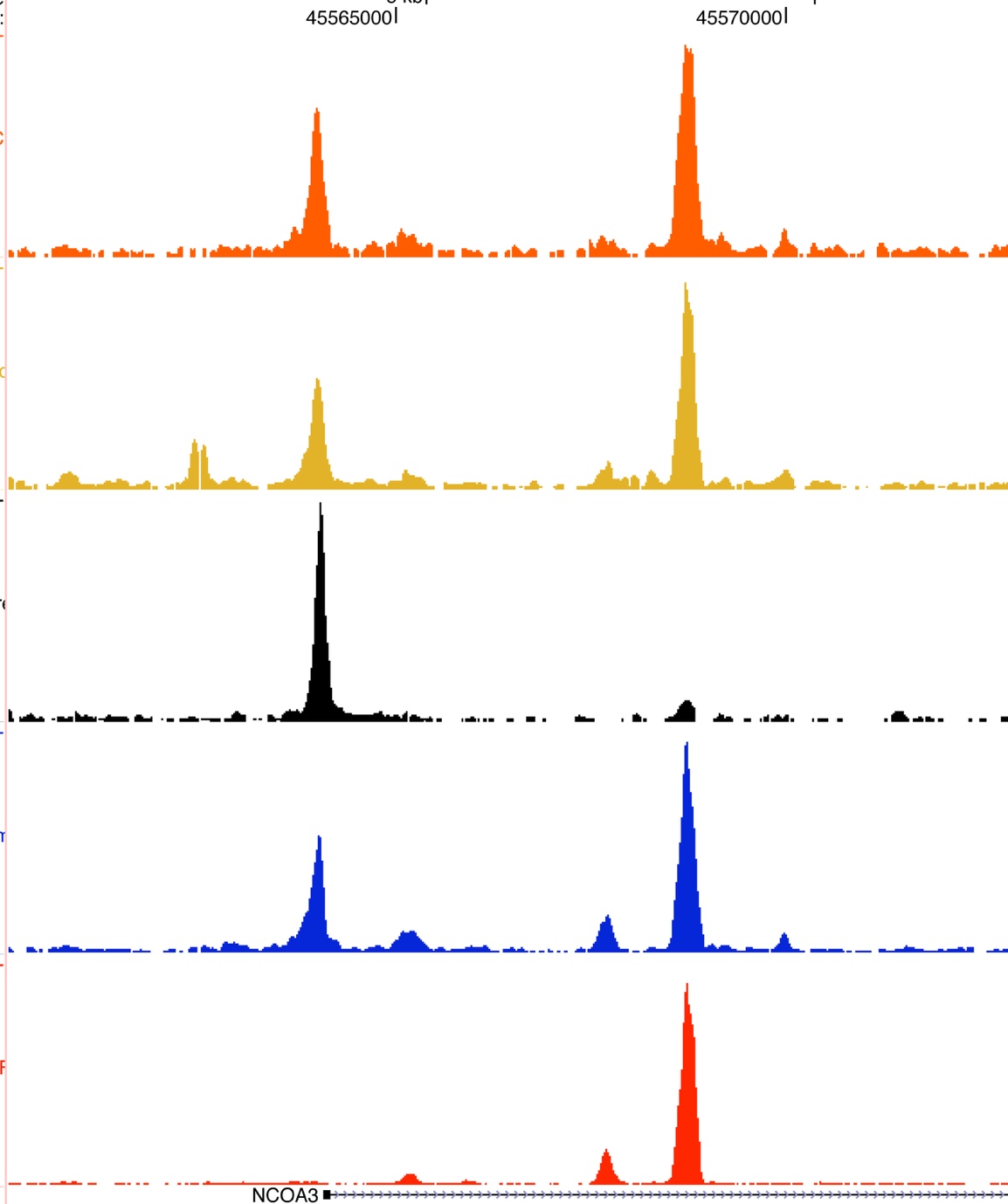
Defining how FOXA1 functions and discovering novel pioneer factors
FOXA1 is the archetypal pioneer factor, where it can bind to compacted chromatin, opening that region of the DNA and subsequently recruiting the COMPASS complex, that initiate the events that ultimately create de novo enhancer elements. As such, FOXA1 is the first step in the creation of enhancers in the contexts where it is expressed, which includes ER+ breast and prostate cancer. Although we know that FOXA1 is an essential gene in these cancers and a biomarker of these tumours, we still do not completely understand how FOXA1 functions and what happens when FOXA1 is altered. This is particularly important given that perturbations in FOXA1 expression and fidelity are commonly observed in advanced disease.
We are interested in defining how FOXA1 associates with the DNA, what proteins are essential for it to function and whether there are post-translational modifications on FOXA1 that alter its activity and might be therapeutically exploited. We take unbiased screening approaches to understand what regulates FOXA1 expression and protein stability and what allows it to mediate COMPASS complex recruitment and therefore enhancer creation. We are identifying amino acid residues on FOXA1 that are important for its ability to be a pioneer factor as well as looking for similarities and differences between its mechanism of action in breast and prostate cancer.
A parallel part of the group is interested in developing new methodological approaches for discovering novel pioneer factors in cancer types where these are not well characterised.
Defining the transcriptional mechanisms in prostate and pancreatic cancer
In prostate cancer, FOXA1 is also a pioneer factor, but in this context is recruits Androgen Receptor (AR). We are interested in defining how FOXA1 and AR co-operate, but our main focus is defining advanced prostate cancer, specifically neuroendocrine prostate cancer (NEPC). NEPC is an aggressive subtype of prostate cancer that we know little about, but it has increased in frequency, in response to more potent tAR-targeted treatments. We are specifically interested in defining the role of FOXA1 in NEPC, as well as discovering NEPC-specific transcription factors and co-factors that are important for this aggressive and difficult to treat cancer subtype.
Recent evidence has suggested that FOXA1 becomes co-opted in pancreatic adenocarcinoma (PDAC), where is creates new enhancer elements that are associated with PDAC metastasis. We focus on characterising the role of FOXA1 in PDAC, with the goal of identifying where it plays a role and how it makes these transcriptional events occur. We are focused on identifying the proteins that FOXA1 works with, in order to identify therapeutic vulnerabilities that we can exploit for pancreatic cancer patients.
Group Members
-

Jason Carroll
Senior Group Leader
-

Ioanna Karouzou
Research Administrator
-

Shalini Rao
Senior Research Associate
-

Soleilmane Omarjee
Senior Research Associate
-

Igor Chernukhin
Bioinformatician
-

Hamish McMillan
Research Associate
-

George Couch
Clinical Fellow
-

Krysia Sadzikowska
Postgraduate Student
-

Catarina Pelicano
Postgraduate Student
-

Josephine Greenall-Ota
Postgraduate Student
-

Marleen Wölke
Postgraduate student
-

Karen Pinilla
Postgraduate student
-

Rebecca Lucey
Visiting Student
-

Fiona Grealy
Research Assistant
-

Rebecca Burrell
Visiting Clinical Lecturer
-

Kristina Kostadinova
Visiting Worker
Related News
See all news-
Hot flush treatment has anti-breast cancer activity, study finds
5th January 2026
A drug mimicking the hormone progesterone has anti-cancer activity when used together with conventional anti-oestrogen treatment for women with breast cancer, a new Cambridge-led trial has found.
Find out more -

Institute scientists uncover molecular switch that drives pancreatic cancer progression
30th October 2025
New research from our Carroll Group has identified a molecular mechanism that helps explain how pancreatic ductal adenocarcinoma progresses, offering a potential path toward more targeted treatments.
Find out more -

Scientists to Shine a Light on Cancer Research at Inspirational Night Walk
10th October 2025
Dr Shalini Rao, a Senior Research Associate at the Cancer Research UK Cambridge Institute, will join postgraduate students Marleen Wolke and Josephine Greenall-Ota for the Shine Night Walk on Saturday, 18 October.
Find out more
Further reading
-

Behind the Lab Coat with Prof Jason Carroll
Read moreGet to know the people who are doing amazing science at the Institute.
-

The PIONEER clinical trial
Read moreThis is a summary of the initial findings from the PIONEER clinical trial which has translated some of the biological findings made in the Carroll lab.
-

Twitter
Find out moreFollow our twitter account to hear all our latest news.
Publications
-
The EstroGene Database Reveals Diverse Temporal, Context-Dependent, and Bidirectional Estrogen Receptor Regulomes in Breast Cancer.
E-pub date: 15 Aug 2023
-
FOXA1 Reprogramming Dictates Retinoid X Receptor Response in ESR1-Mutant Breast Cancer.
E-pub date: 1 Jun 2023
-
Peroxide-cleavable linkers for antibody-drug conjugates.
E-pub date: 9 Feb 2023
-
Identification of a novel GR-ARID1a-P53BP1 protein complex involved in DNA damage repair and cell cycle regulation.
E-pub date: 1 Dec 2022
Laboratory Efficiency Assessment Framework (LEAF)
The Carroll Group contributed to the Institute’s LEAF Silver accreditation, see the Sustainability webpage for more information.