Narita Group
Cellular senescence and tumour suppressors

Research Summary
We study how stress affects our cells, causing them to age and stop working properly. Our DNA, which contains our genetic information, can get damaged over time due to aging or UV light, making cells stop functioning as they should. These aged cells, known as senescent cells, no longer help repair our bodies. We aim to understand how this process works, including how DNA organisation affects cell changes, how certain genes linked to cancer cause cells to age, and how the buildup of these aged cells over time leads to aging.
Introduction
Senescence is a state of persistent cell cycle arrest triggered by various stimuli but senescent are not inert. Rather they actively communicate with their surroundings, shaping the tissue microenvironment and potentially burdening the individual, especially in aging but also in cancer. We are particularly interested in understanding what senescent cells do to tissues and how they achieve such altered functionality.

Professor Masashi Narita
Senior Group Leader
Focus areas
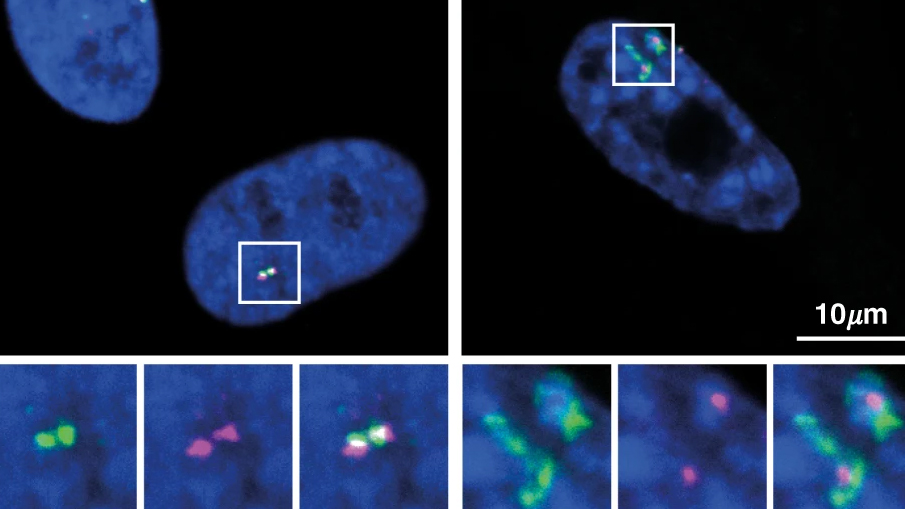
High-order chromatin organisation
Our group studies the global chromatin reorganisation which often accompanies senescence, marked by the formation of senescence-associated heterochromatin foci (SAHFs, Narita et al. Cell 2003). The senescence model we focus on is oncogene-induced senescence (OIS), though we often include comparisons to replicative and DNA-damage induced senescence. SAHF formation is a multifactorial process involving the High Mobility Group A (HMGA) proteins and disruption of the Lamin B1 chromatin tethering at the nuclear periphery. During SAHF-formation, chromatin is organised into concentric epigenetic layers with H3K9m3-core with H3K27me3-shell, excluding euchromatin regions (Chandra et al. Mol Cell 2012). See our recent review article on this topic (Olan, Handa and Narita, Curr. Opinion in Cell Biol. 2023). Recently, we showed that HMGA1 affects chromatin organisation beyond its effect on SAHF, strengthening compartmentalisation (Olan, Ando-Kuri and Parry et al. 2024, Research Square Preprint). HMGA1 mobilises chromatin depending on the levels of HMGA1 binding, leading to gene repositioning corelated with up- and down-regulation of key senescence genes. Future directions include elucidating nuances of the function of HMGA1 towards chromatin organisation and gene regulation depending on protein modifications and protein binding partners.
Chromatin reorganisation during senescence is also associated with SAHF-dependent desilencing of ‘lineage-inappropriate’ genes from H3K9me3 heterochromatin primarily at peri-SAHF regions (Tomimatsu et al. Nat Aging 2022), raising important questions about the shift in cell identity. The genes contributing to the senescence-associated secretory phenotype (SASP) often show tissue specificity. See our recent review article on this topic (Olan and Narita. Annu. Rev. Cell Dev. Biol. 2022). SASP is also supported by chromatin loops rewiring due to loss cohesin binding as well as the formation of transcription-dependent cohesin islands (Olan et al. Nat Comm 2020). Our future work aims to integrate the chromatin changes observed during senescence from small scale (loops and enhancer-promoter interactions) to large scale organisation between domains located megabases apart on the linear genome.
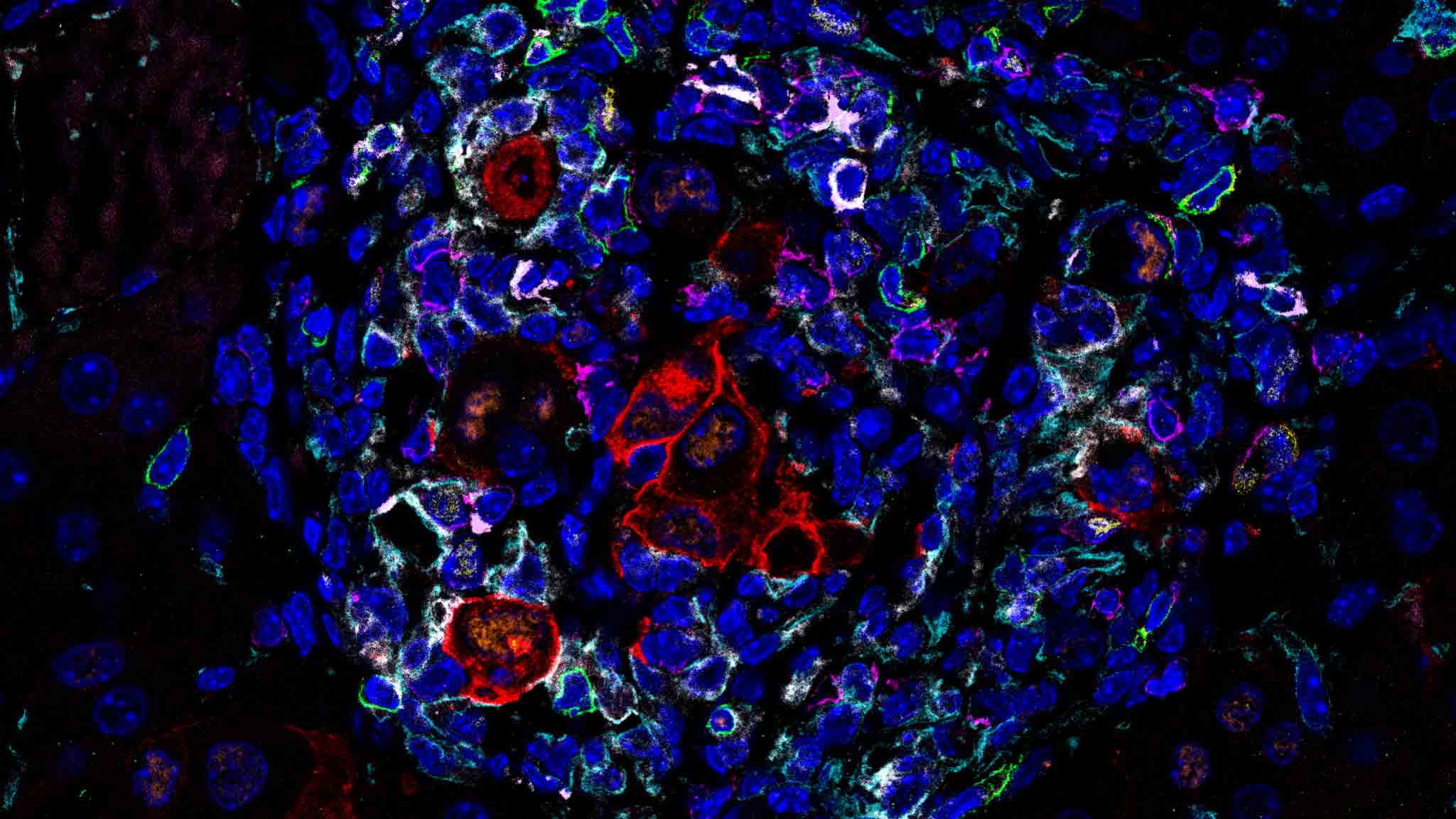
Titration of oncogenic RAS
Our RAS dose-titrating systems model a non-linear oncogene-induced senescence (OIS) spectrum, including senescence-intermediates such as immune-resistant tumour initiating states, characterised by increased progenitor features and a reduced Myc signature. Moreover, our study uncovers a RAS-dose-associated evolution of senescence and its immune-microenvironment, revealing at least two distinct paths towards tumorigenesis in the liver: 1) a Dlk1/Afp-branch, corresponding to differentiated HCCs with longer latency and 2) a Notch1/Tgfb1/Nes-branch, corresponding to undifferentiated tumours, associated with short latency and poor prognosis.
Chan et al. 2024. Nature. In Press.
(Pre-print: https://doi.org/10.21203/rs.3.rs-2842963/v1)
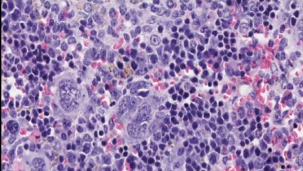
Age-associated spontaneous cancer mouse model
We have shown the functional relevance of autophagy in senescence (Young, Narita et al. Genes Dev 2009, Narita, Young et al. Science 2011). To further extend these in vitro studies, we have generated RNAi based autophagy mouse models, where we can switch on/off autophagy using Doxycycline (Dox) -inducible sh-Atg5 (Cassidy, Young, Pérez-Mancera et al. Autophagy 2018). They are hypomorphic and also allow for temporospatial regulation of autophagy without any confounding effects on development. We genetically validated a long-standing question: in mammals does the autophagy-decline induce premature ageing? Indeed, this was the case. Moreover, restoration of autophagy led to a dramatic age reversal, however this was associated with a dramatic increase in tumorigenesis (Cassidy et al. Nat Commun 2020). Our model therefore provides a platform for investigating age-associated tumorigenesis. See our review article on this topic (Cassidy and Narita. Mol. Oncol. 2022)
Group members
-

Masashi Narita
Group Leader
-

Masako Narita
Principal Scientific Associate
-

Andrew Young
Principal Scientific Associate
-

Adelyne Chan
Research Associate
-

Ioana Olan
Research Associate
-

Yongmin Kwon
Postgraduate Student
-

Tetsuya Handa
Postgraduate Student
-

Haoran Zhu
Research Associate
-

George Skalka-Hall
Research Associate
Related News
See all news-
Targeting paused cells could improve chemotherapy for lung and ovarian cancers
3rd February 2026
New research published today in Nature Aging by scientists at the University of Cambridge sheds light on why some lung and ovarian cancers stop responding to chemotherapy, and how this resistance might one day be prevented.
Find out more -

Aleksandra Janowska awarded Postgraduate Student Thesis Prize
25th November 2025
Aleksandra Janowska has won this year’s Postgraduate Student Thesis Prize. The Prize is awarded each year to a student who has undertaken an outstanding project to the highest standards during the course of their PhD study.
Find out more -
Mutations in liver cells linked to liver disease and fat metabolism
13th October 2021
Research from the Narita Group and their collaborators has identified mutations linking liver disease with obesity and diabetes, leading to new understanding about how systemic diseases interact.
Find out more
Publications
See all publications-
Senescence-induced endothelial phenotypes underpin immune-mediated senescence surveillance.
Narita Group
E-pub date: 1 May 2022
-
Locus-specific induction of gene expression from heterochromatin loci during cellular senescence
Narita Group
E-pub date: 28 Dec 2020
-
Transcription-dependent cohesin repositioning rewires chromatin loops in cellular senescence.
Narita Group
E-pub date: 27 Nov 2020
-
Temporal inhibition of autophagy reveals segmental reversal of ageing with increased cancer risk.
Narita Group
E-pub date: 16 Jan 2020
Laboratory Efficiency Assessment Framework (LEAF)
The Narita Group contributed to the Institute’s LEAF Silver accreditation, see the Sustainability webpage for more information.