Balasubramanian Group
Chemical biology

Research summary
We investigate special DNA structures called quadruplexes that are involved in the expression of cancer genes. Using chemical biology methods, we have invented small drug-like molecules that bind to these structures, stabilising them. This can switch off cancer genes and thus stop cancers growing. These drug-like molecules may work on many different cancer genes making them potentially much less toxic for non-cancer cells than conventional drugs.
Introduction
Recent advances in understanding nucleic acid function have shown that alternative secondary structures and the chemical modification of nucleotide bases have key regulatory roles in diverse cellular processes, from transcription and translation to cell division and genome stability.
Genetic information is carried not only by the sequence of nucleic acids, but also their secondary structures and chemical modifications. For example, guanine-rich sequences can form stable four‑stranded structures called G-quadruplexes (G4s), while certain cytosine bases in DNA can become methylated. We hypothesise that such alternative structures, or chemical modifications, have critical functions in normal cells and cancer. By identifying where base modifications and G4 structures are located in the cancer cell genome, and through the application of synthetic small molecules that selectively target G4 structures, we aim to understand the oncogenic process and develop novel approaches for potential use in treatment and diagnosis of cancer. We are also exploring new strategies to target the DNA binding activity of FOXM1, a key cancer-related transcription factor involved in cell cycle control.

Professor Sir Shankar Balasubramanian
Senior Group Leader
Research areas
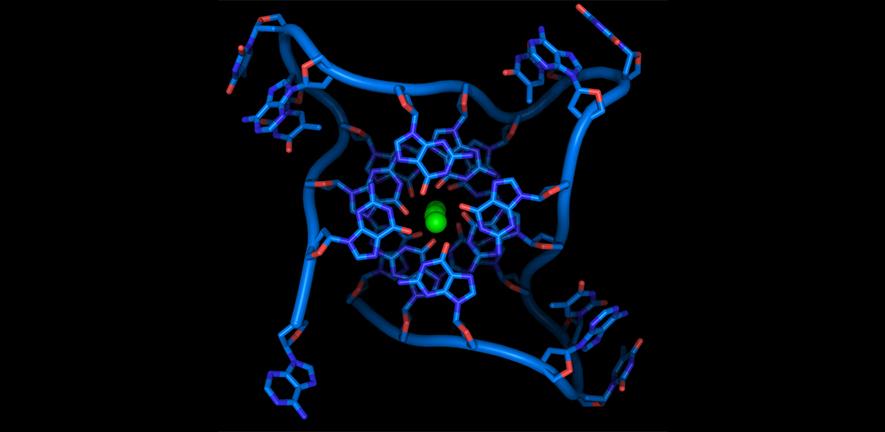
G quadruplexes
Nucleic acids are highly flexible molecules than can adopt different conformational structures. While in living systems DNA is largely double helical and RNA is single stranded, guanine-rich sequences can exist in alternative structural forms known as G-quadruplex nucleic acids. We are investigating the functional relevance of a quadruple helical form of nucleic acids and its implication for the biology of nucleic acids. The G-quadruplex hypothesis is of fundamental importance to life and may well hold the key to new therapeutic approaches in numerous areas of human disease that include cancer.
Specific mechanisms under investigation include: DNA G-quadruplex formation at the telomeres and their importance for genomic stability and replication; DNA G-quadruplexes in gene promoters and the regulation of transcription; RNA G-quadruplexes in the untranslated regions of mRNA and the control of protein synthesis (translation).
As part of our studies, we are synthesising new molecules that stabilise the nucleic acid quadruple helix and interfere with specific cellular processes. We make extensive use of biophysical methods (NMR, UV, CD and fluorescence spectroscopy) to study quadruplexes and their interactions with molecules. We are also employing computational methods (bioinformatics) and genomics to explore quadruplexes in genomes.

Epigenetics and Modified Bases
We are interested in understanding chemical modifications to DNA and the effect of such changes to the structure and function of DNA. DNA is made up of four bases – cytosine, guanine, adenine and thymine. However, these bases can naturally undergo chemical modification leading to new bases. Changing one of the bases in a strand of DNA in this way alters its property and function by controlling how the sequence is interpreted. This can affect how genes are switched on and off in different cell types, tissues and organs.
The modified base 5-methylcytosine (5mC) is a well-known epigenetic mark that can regulate transcription of the genome. Since 2009 three further modified bases have been detected in the mammalian genome. These are the TET-enzyme generated bases; 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). The presence of these modifications opens up questions as to their function in normal cellular biology and disease states.
We are developing chemical tools and genomic methods to map and elucidate the function of these modified bases.
We are also exploring the molecular basis for their involvement in biological mechanisms. Part of this work exploits state of the art genomics technologies. We have already created methods to quantitatively sequence 5mC, 5hmC and 5fC at single-base resolution. Such tools allow much more accurate study of these epigenetic marks.
The scope of our work will also include the identification, mapping, and elucidation of the biological function of other base modifications in the DNA and RNA of various organisms.
Related News
See all news-

Prof Sir Shankar Balasubramanian Awarded Prestigious Royal Society of Chemistry Prize
25th June 2025
Professor Sir Shankar Balasubramanian has received the Royal Society of Chemistry Khorana Prize, awarded for outstanding contributions through work at the chemistry and life science interface.
Read more -
Sir Shankar Balasubramanian elected Fellow of the AACR Academy
10th March 2025
Sir Shankar has been recognised for his pioneering research in nucleic acid chemistry and his key role in the development of next-generation sequencing technology.
Find out more -

Isabel Esain Garcia awarded PhD Thesis Prize
3rd December 2024
The Prize is awarded annually to a student who has undertaken an outstanding research project to the highest standards during the course of their PhD study at the Cancer Research UK Cambridge Institute.
Find out more
Publications
-
Improved simultaneous mapping of epigenetic features and 3D chromatin structure via ViCAR.
E-pub date: 27 Nov 2024
-
Spatially tuneable multi-omics sequencing using light-driven combinatorial barcoding of molecules in tissues
E-pub date: 25 May 2024
-
An Upstream G-Quadruplex DNA Structure Can Stimulate Gene Transcription.
Balasubramanian Group
E-pub date: 15 Mar 2024
-
G-quadruplex DNA structure is a positive regulator of MYC transcription.
E-pub date: 13 Feb 2024
Laboratory Efficiency Assessment Framework (LEAF)
The Balasubramanian Group contributed to the Institute’s LEAF Silver accreditation, see the Sustainability webpage for more information.